How Motif Matching Works
This vignette explains what happens when you call
have_motif() or count_motif().
In this vignette, we describe the matching rules that power all
glymotif functions. The rules are rooted in the
comprehensive GlycoMotif
database, and adapted for practical glycan analysis workflows.
A quick note: We use IUPAC-condensed glycan text representations throughout. If this format is unfamiliar, start with this primer.
Defining Our Terms
Before diving into the technical details, let’s establish some clarity about what we’re actually matching.
Throughout this vignette, “glycan” refers to a complete carbohydrate structure—the whole molecular tree, from its reducing end (often attached to proteins or lipids) to its non-reducing termini.
“Motif”, on the other hand, is any structurally meaningful pattern within that tree. It could be:
- A single monosaccharide
- A small oligosaccharide unit
- An entire glycan structure
Our central question is simple: Does the given glycan contain this particular motif?
Let’s start with a visual example that illustrates this concept:
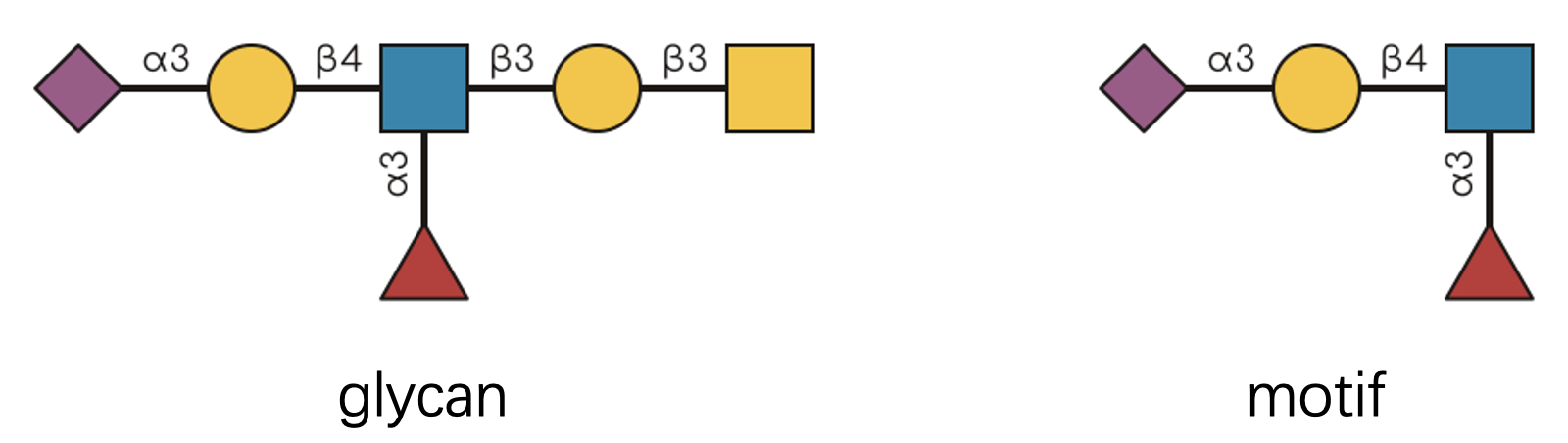
Looking at this diagram, we can confidently say “the glycan contains
the motif with exactly 1 occurrence.” The presence part is determined by
have_motif(), while the counting aspect is handled by
count_motif().
glycan <- "Neu5Ac(a2-3)Gal(b1-3)[Fuc(a1-6)]GlcNAc(b1-3)Gal(b1-3)GalNAc(b1-"
motif <- "Neu5Ac(a2-3)Gal(b1-3)[Fuc(a1-6)]GlcNAc(b1-"
print(paste0("Does the glycan have the motif? ", have_motif(glycan, motif)))
#> [1] "Does the glycan have the motif? TRUE"
print(paste0("How many occurrences of the motif are there in the glycan? ", count_motif(glycan, motif)))
#> [1] "How many occurrences of the motif are there in the glycan? 1"Why Not Just Use str_detect()?
You might be thinking: “This example looks straightforward—why not just use string matching?” Let’s test that hypothesis:
stringr::str_detect(glycan, stringr::fixed(motif))
#> [1] TRUEIndeed, it works for this simple case. But string matching does not handle the common complications in glycan data.
Real-world glycan analysis is often complex. Consider these challenging scenarios:
- Complex branching patterns with multiple attachment points
- Ambiguous linkage annotations where details are missing or uncertain
- Generic monosaccharide assignments from mass spectrometry data
- Chemical modifications and substituents that add complexity
- Positional constraints where context determines biological meaning
- Reducing end anomers that affect molecular recognition
Writing regular expressions to handle all these nuances? That becomes
difficult to maintain quickly. That is why glymotif uses
glycan-aware matching rules.
Matching Rules
The have_motifs() and count_motifs()
functions return matrices with meaningful row and column names. For
clarity in our demonstrations, let’s create simplified wrapper
functions:
# You don't have to understand this.
have_motifs_simple <- function(glycan, motifs, ...) {
unname(have_motifs(glycan, motifs, ...)[1, ])
}
count_motifs_simple <- function(glycan, motifs, ...) {
unname(count_motifs(glycan, motifs, ...)[1, ])
}Now, let’s explore each matching rule systematically.
Rule 1: Branching Logic
Branching patterns are easier to reason about when glycans are treated as tree structures. Let’s examine this with a concrete example:
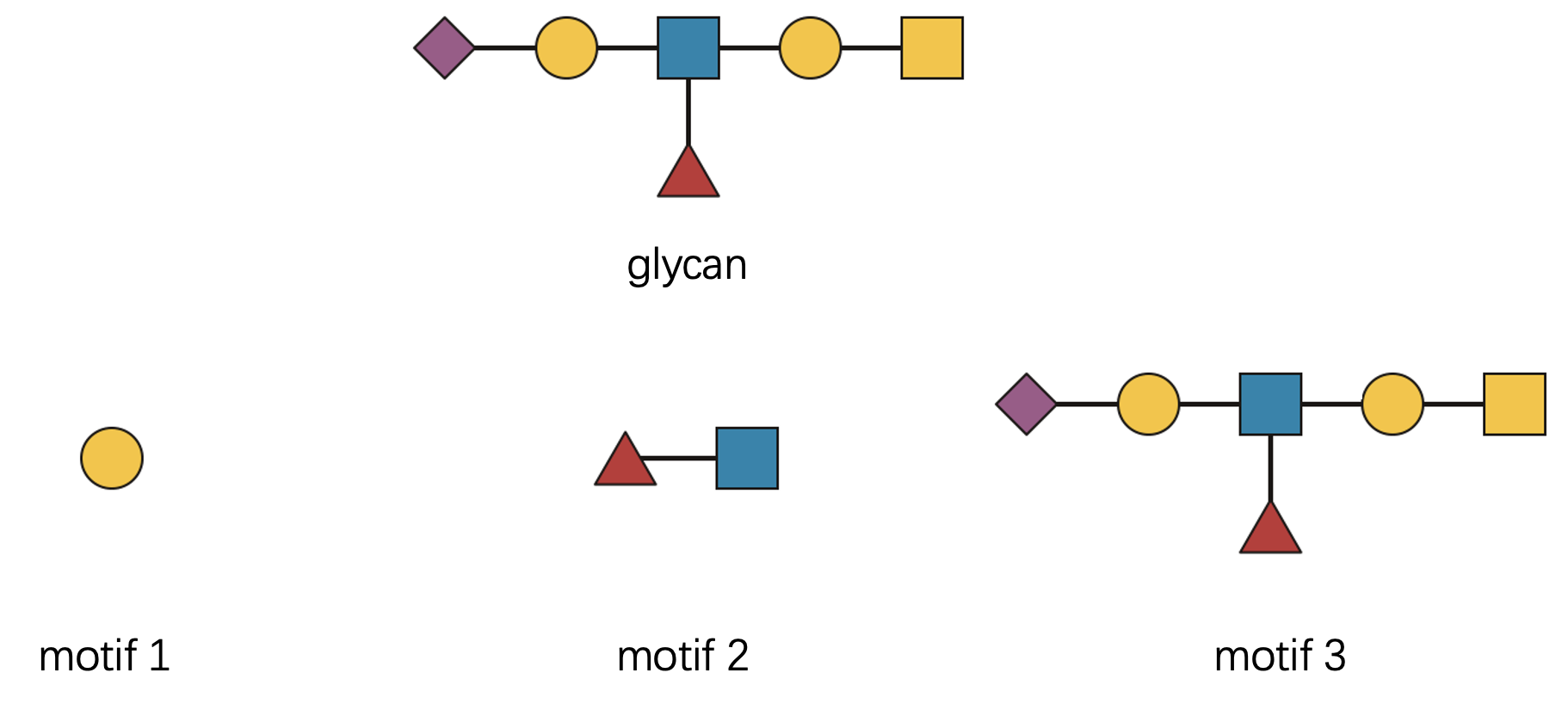
Let’s check three distinct motifs, each representing a different level of structural complexity:
- “Gal(??-” - a single monosaccharide building block
- “Fuc(??-?)GlcNAc(??-” - a disaccharide with ambiguous linkage
- The complete glycan structure itself
glycan <- "Neu5Ac(??-?)Gal(??-?)[Fuc(??-?)]GlcNAc(??-?)Gal(??-?)GalNAc(b1-"
motifs <- c(
"Gal(??-",
"Fuc(??-?)GlcNAc(??-",
glycan
)
count_motifs_simple(glycan, motifs)
#> [1] 2 1 1The computational perspective: Behind the scenes, we’re performing subgraph isomorphism matching. Glycans and motifs are represented as mathematical graphs, and we’re searching for structural embeddings.
But there are two distinctions from standard graph theory:
First, directionality matters. The reducing end (right side) and non-reducing end (left side) are biologically distinct. Direction affects function:
motifs <- c("Fuc(??-?)GlcNAc(??-", "GlcNAc(??-?)Fuc(??-")
have_motifs_simple(glycan, motifs)
#> [1] TRUE FALSESecond, biological equivalence matters more than mathematical multiplicity. When multiple mathematically distinct matches have identical biological meaning, we count them as one.
Consider this example:
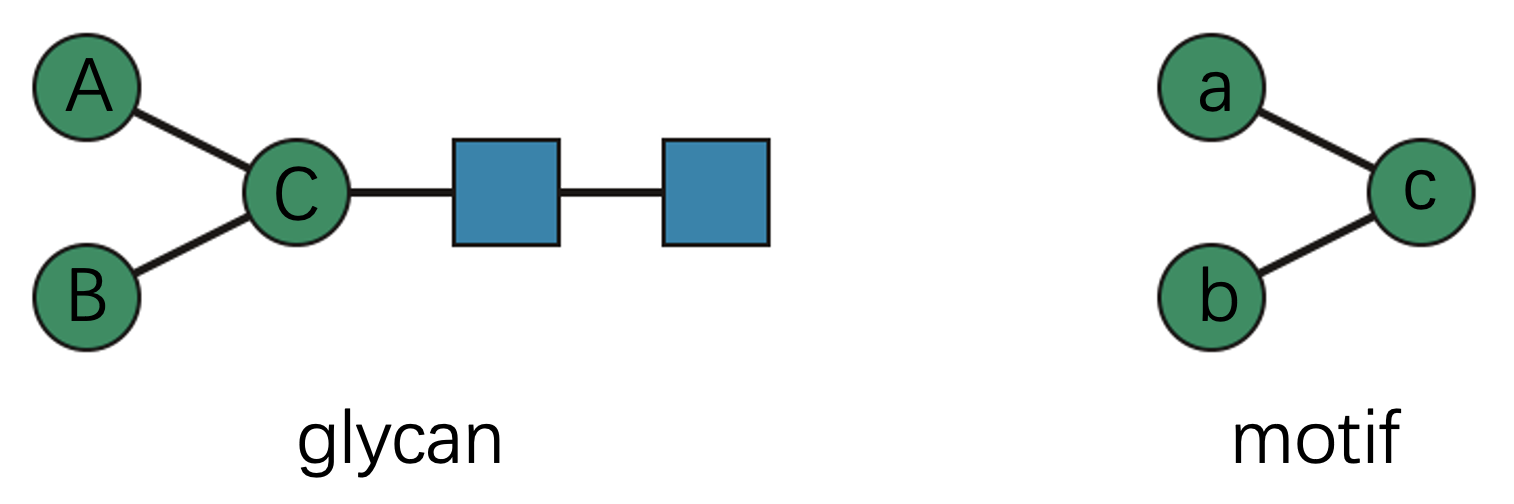
Technically, this motif has two valid subgraph matches within the
glycan (“A-a, B-b, C-c” and “A-b, B-a, C-c”). But from a biological
perspective, these matches are equivalent—the specific assignment of
mannose residues doesn’t matter. Therefore, count_motif()
reports exactly one match:
glycan <- "Man(??-?)[Man(??-?)]Man(??-?)GlcNAc(??-?)GlcNAc(??-"
motif <- "Man(??-?)[Man(??-?)]Man(??-"
count_motif(glycan, motif)
#> [1] 1Rule 2: Linkage Flexibility
Linkage information in glycomics is often incomplete. You might encounter patterns like “??-6”, “a2-?”, or complete unknowns. The matching rule is:
The glycan cannot be more ambiguous than the motif.
This means a concrete linkage like “a2-6” in your glycan data will match:
- “a2-6” (exact match)
- “a2-?” (position-specific, anomer flexible)
- “??-6” (anomer-specific, position flexible)
- “??-?” (completely flexible wildcard)
But an ambiguous linkage like “a2-?” will only match equally or more flexible patterns in the motif.
Let’s see this in practice:
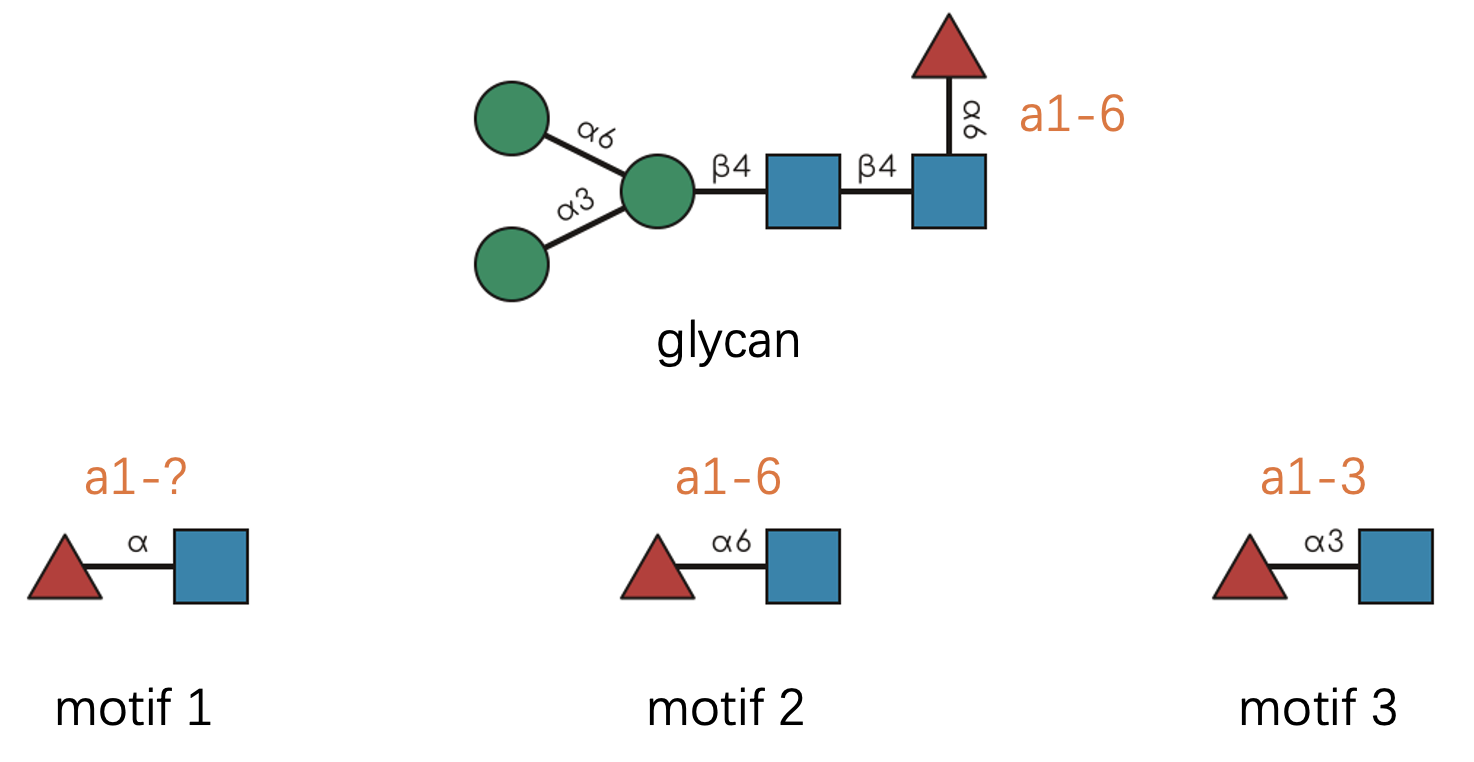
A note about notation: Following SNFG conventions, we often abbreviate linkages by omitting the anomeric carbon number. So “a1-6” becomes simply “a6” since the anomeric position is typically known.
glycan <- "Man(a1-3)[Man(a1-6)]Man(b1-4)GlcNAc(b1-4)[Fuc(a1-6)]GlcNAc(b1-"
motifs <- c(
"Fuc(a1-?)GlcNAc(b1-", # Motif 1: anomer known, position flexible
"Fuc(a1-6)GlcNAc(b1-", # Motif 2: exact linkage match
"Fuc(a1-3)GlcNAc(b1-" # Motif 3: wrong position specification
)
have_motifs_simple(glycan, motifs)
#> [1] TRUE TRUE FALSERule 3: Monosaccharide Resolution
Mass spectrometry often provides incomplete monosaccharide identification. You might know there’s a hexose present but not whether it’s glucose, galactose, or mannose.
We distinguish between two resolution levels:
- Concrete monosaccharides: Structurally specific (e.g., “Gal”, “Man”, “Glc”)
- Generic monosaccharides: Compositionally defined (e.g., “Hex”, “HexNAc”, “dHex”)
The matching rule mirrors our linkage philosophy: The glycan cannot be more ambiguous than the motif.
Specifically:
- Concrete monosaccharides in glycans can match both concrete and generic motifs
- Generic monosaccharides in glycans can only match generic motifs
have_motif("Gal(a1-", "Gal(a1-")
#> [1] TRUE
have_motif("Gal(a1-", "Hex(a1-")
#> [1] TRUE
have_motif("Hex(a1-", "Gal(a1-")
#> Warning: Matching lower-level `glycans` against higher-level `motifs` usually returns no
#> matches.
#> ℹ `glycans` have "basic" structure level, while `motifs` have "intact"
#> structure level.
#> ℹ Use motifs at the same structure level as the glycans, or reduce motif
#> structure levels before matching.
#> ℹ See `?get_structure_level` for details.
#> [1] FALSE
have_motif("Hex(a1-", "Hex(a1-")
#> [1] TRUERule 4: Chemical Modifications
Real glycans are often decorated with chemical modifications—acetylation, sulfation, methylation, and more. These substituents have two components: position (where they’re attached) and identity (what they are).
For example, “Neu5Ac9Ac” represents N-acetylneuraminic acid with an additional 9-O-acetyl group.
The matching rules are:
- Identity matching: If the glycan has a substituent, the motif must have the same type to match
- Position flexibility: The glycan cannot be more ambiguous than the motif regarding position
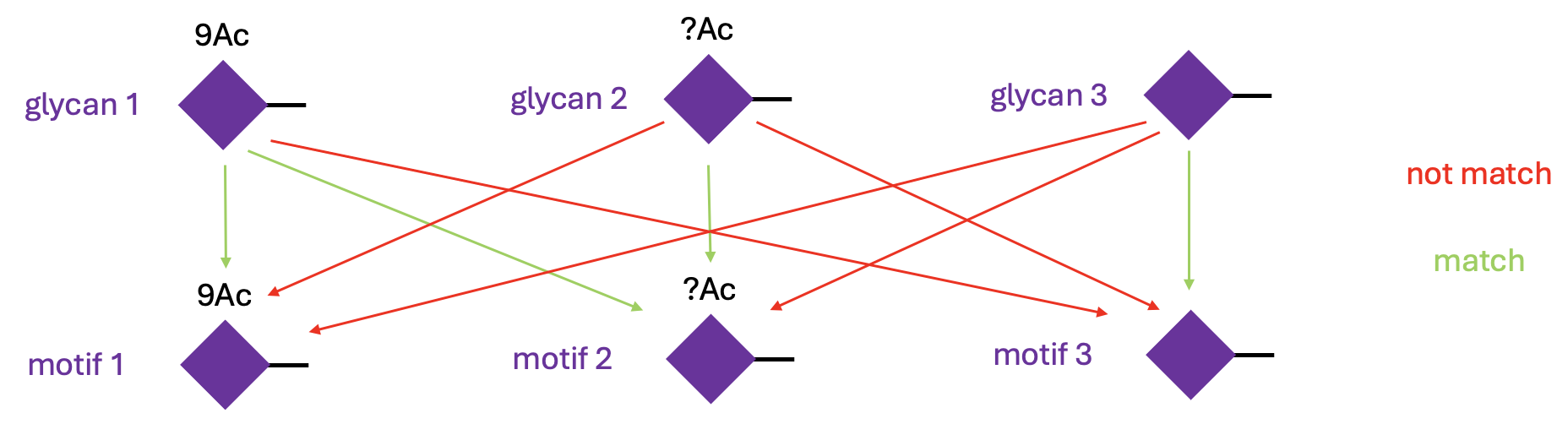
Let’s see this in action:
glycans <- c("Neu5Ac9Ac(a2-", "Neu5Ac?Ac(a2-", "Neu5Ac(a2-")
motifs <- c("Neu5Ac9Ac(a2-", "Neu5Ac?Ac(a2-", "Neu5Ac(a2-")
mat <- have_motifs(glycans, motifs)
rownames(mat) <- paste0("glycan_", 1:3)
colnames(mat) <- paste0("motif_", 1:3)
mat
#> motif_1 motif_2 motif_3
#> glycan_1 TRUE TRUE FALSE
#> glycan_2 FALSE TRUE FALSE
#> glycan_3 FALSE FALSE TRUEThe default behavior is to match the substituent strictly. This is
reasonable in most cases, because monosaccharides with different
substituents should be regarded as different. However, you can change
this behavior by setting strict_sub = FALSE. In this case,
the substituent is optional in the motif, so the glycan “Neu5Ac9Ac” can
match the motif “Neu5Ac”.
have_motif("Neu5Ac9Ac(a2-", "Neu5Ac(a2-", strict_sub = FALSE)
#> [1] TRUERule 5: Alignment Constraints
Some motifs are only meaningful in specific structural contexts.
Consider the N-glycan core—it’s biologically significant only when positioned at the reducing end. Similarly, the Tn antigen (simply GalNAc) should represent the entire O-glycan structure, not just any GalNAc residue buried within a larger molecule.
Following GlycoMotif standards, we recognize four alignment types:
- “substructure”: The motif can appear anywhere within the glycan
- “core”: Must align with a connected subtree at the reducing end
- “terminal”: Must align with a connected subtree at non-reducing ends
- “whole”: Must match the entire glycan structure
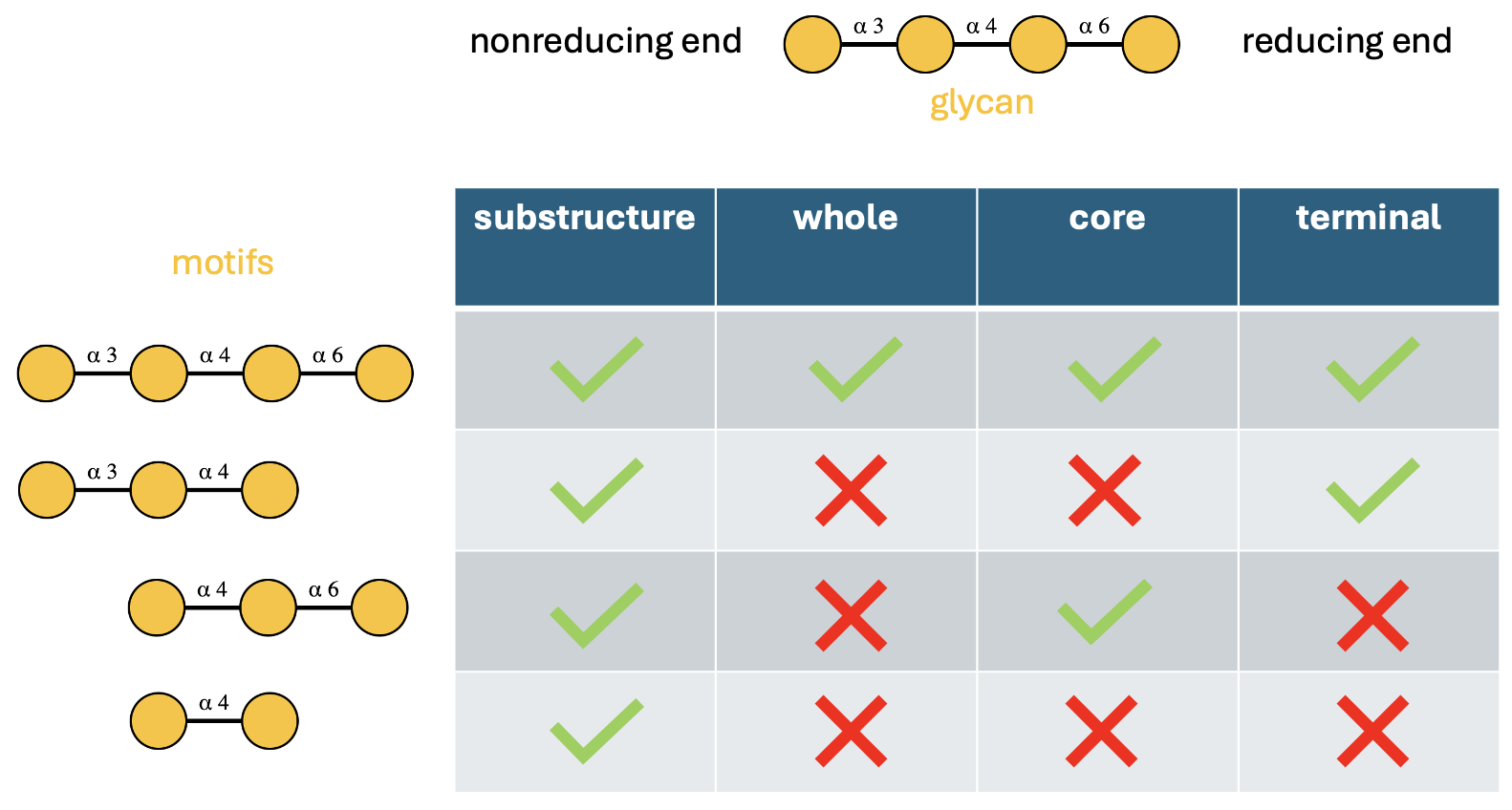
Let’s verify these behaviors computationally:
glycan <- "Gal(a1-3)Gal(a1-4)Gal(a1-6)Gal(a1-"
motifs <- c(
"Gal(a1-3)Gal(a1-4)Gal(a1-6)Gal(a1-", # motif 1: complete structure
"Gal(a1-3)Gal(a1-4)Gal(a1-", # motif 2: terminal branch
"Gal(a1-4)Gal(a1-6)Gal(a1-", # motif 3: reducing-end subtree
"Gal(a1-4)Gal(a1-" # motif 4: internal fragment
)
alignments <- c("substructure", "whole", "core", "terminal")
mat <- do.call(cbind, purrr::map(alignments, ~ have_motifs_simple(glycan, motifs, alignment = .x)))
colnames(mat) <- alignments
rownames(mat) <- paste0("motif_", 1:4)
mat
#> substructure whole core terminal
#> motif_1 TRUE TRUE TRUE TRUE
#> motif_2 TRUE FALSE FALSE TRUE
#> motif_3 TRUE FALSE TRUE FALSE
#> motif_4 TRUE FALSE FALSE FALSERule 6: Reducing End Anomers
The reducing end of a glycan—that special monosaccharide connected to proteins or lipids—deserves special attention. Its anomeric configuration can significantly impact biological function.
The matching behavior depends on motif alignment:
When the motif aligns away from the reducing end: The motif’s “reducing end” (really just its rightmost residue) is matched against the corresponding internal linkage.
glycan <- "Gal(a1-3)GalNAc(b1-"
motifs <- c("Gal(a1-", "Gal(b1-")
have_motifs_simple(glycan, motifs)
#> [1] TRUE FALSEWhen the motif aligns at the reducing end: Direct comparison with the glycan’s actual reducing end anomer.
glycan <- "Gal(a1-3)GalNAc(b1-"
motifs <- c("GalNAc(a1-", "GalNAc(b1-")
have_motifs_simple(glycan, motifs)
#> [1] FALSE TRUEWhy This Complexity Matters
You might be wondering: “Why all these intricate rules?” The answer lies in the complexity of biological systems.
Unlike artificial pattern matching, biological recognition systems are:
- Context-sensitive: The same motif can have different meanings in different locations
-
Fault-tolerant: Partial information should still
yield meaningful results
- Hierarchically organized: Generic patterns can be refined into specific ones
- Chemically aware: Modifications and substitutions are integral to function
By encoding these biological principles into our matching algorithms,
glymotif bridges the gap between computational analysis and
biological reality.
Whether you’re analyzing clinical glycomics data, exploring evolutionary relationships, or designing glycan-based therapeutics, these matching rules help keep results both computationally sound and biologically meaningful.
Ready for More?
These motif matching rules provide the foundation for understanding
how glymotif works. With this knowledge, you can interpret
motif matching results in more complex glycan analysis workflows.
For practical applications and real-world examples, head back to the Getting Started guide. For detailed function documentation, explore the reference manual.
This should give you a practical basis for using the matching functions in your own analyses.